![]() .
.
Величина ![]() описывает
изменение энергии Гиббса одного моля растворяемого газообразного вещества при
переходе из газовой фазы в жидкую.
описывает
изменение энергии Гиббса одного моля растворяемого газообразного вещества при
переходе из газовой фазы в жидкую.
9.4. Предельно разбавленные растворы
В очень разбавленных растворах исчезает взаимодействие между молекулами растворенного вещества (но, естественно, не между молекулами растворителя и растворенного компонента). Поэтому можно полагать, что все растворы при сильном разбавлении становятся идеальными по отношению к растворенному соединению.
Интересно установить характер зависимостей термодинамических величин от молярной доли компонента, присутствующего в системе в малом количестве (х2). Из обобщенного уравнения Гиббса-Дюгема (5.35) при постоянных р и Т для i = 1, 2 (бинарная смесь) следует
![]() .
.
Откуда
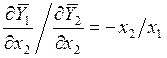 , (9.9)
, (9.9)
где ![]() и
и ![]() - любые парциальные молярные величины.
Видно, что при
- любые парциальные молярные величины.
Видно, что при ![]()
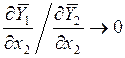 .
.
Для выполнения последнего соотношения необходимо
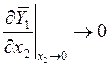 и
и 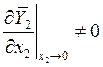 (9.10)
(9.10)
или
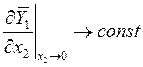 и
и 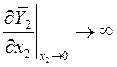 . (9.11)
. (9.11)
Рассмотрим, какие термодинамические величины удовлетворяют соотношениями (9.10), а какие (9.11). Для этого обратимся к идеальным двухкомпонентным системам.
Величины парциальных энергий Гиббса определяются соотношениями
![]() ,
, ![]() . (9.12)
. (9.12)
Дифференцируя оба уравнения по х2 и устремляя к пределу при х2 → 0, получаем
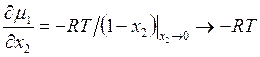 , (9.13)
, (9.13)
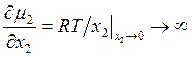 . (9.14)
. (9.14)
Итак, μ1 и μ2 подчиняются соотношению (9.11). При малых значениях х2 для μ1 и μ2 будем иметь
![]() ,
, ![]() . (9.15)
. (9.15)
Рассмотрим энтропию. Учитывая (9.12), можно записать
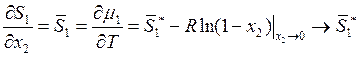 , (9.16)
, (9.16)
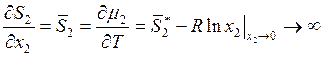 , (9.17)
, (9.17)
где ![]() и
и ![]() - энтропии чистых жидкостей.
Следовательно, энтропия также относится к случаю, описываемому уравнениями
(9.11). При малых значениях х2 в первом приближении
- энтропии чистых жидкостей.
Следовательно, энтропия также относится к случаю, описываемому уравнениями
(9.11). При малых значениях х2 в первом приближении
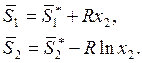 (9.18)
(9.18)
Реально, как
показывают эксперименты, растворы неэлектролитов можно считать идеальными, если
![]() , а в случае изотоп-замещенных молекул
практически при любом значении х2. Что касается растворов
электролитов в воде, то для них условия идеальности выполнены лишь при
, а в случае изотоп-замещенных молекул
практически при любом значении х2. Что касается растворов
электролитов в воде, то для них условия идеальности выполнены лишь при ![]() .
.
В случае
энтальпии уравнение (9.9) неприменимо к идеальным растворам, т.к. величины ![]() и
и ![]() не
зависят от х2. В неидеальных системах величина
не
зависят от х2. В неидеальных системах величина ![]() - энтальпии растворения при бесконечном
разбавлении. Величина
- энтальпии растворения при бесконечном
разбавлении. Величина ![]() , т.к. малые добавки
второго компонента не сказываются на величине
, т.к. малые добавки
второго компонента не сказываются на величине ![]() .
Следовательно, энтальпия удовлетворяет соотношению (9.10). Аналогичным образом
можно рассмотреть и другие величины.
.
Следовательно, энтальпия удовлетворяет соотношению (9.10). Аналогичным образом
можно рассмотреть и другие величины.
9.5. Фазовые диаграммы двухкомпонентных жидких растворов. Равновесие жидкость – пар. Полная смешиваемость в обеих фазах
Для наглядного отображения свойств жидкости и пара в бинарных смесях обычно строят диаграммы трех типов (рис. 9.1):
Необходимо отметить, что координату х2 обычно приписывают веществу с более высоким давлением пара или с более низкой температурой кипения. Для диаграмм первых двух типов вдоль оси абсцисс откладывают одновременно молярную долю второго вещества в паре - х2п и в жидкости - х2ж.
На диаграммах температура – состав кривая пара Т(х2п) описывает зависимость равновесной температуры от состава пара при фиксированном давлении пара, а кривая жидкости Т(х2ж) описывает зависимость равновесной температуры системы от состава жидкой фазы при том же постоянном значении давления пара. В этом случае кривая пара проходит выше кривой жидкости.
На диаграммах давление – состав кривая жидкости р(х2ж) описывает зависимость равновесного давления пара от состава жидкости при фиксированной температуре всей системы, а кривая пара р(х2п) описывает зависимость равновесного давления пара от состава паровой фазы при той же фиксированной температуре. В этом случае, наоборот, кривая жидкости лежит выше кривой пара.
Уважаемый посетитель!
Чтобы распечатать файл, скачайте его (в формате Word).
Ссылка на скачивание - внизу страницы.