mA + mB = m0 = const (10)
Частицы А и В могут быть независимыми, и тогда речь идёт об анализе специальных смесей, либо эти частицы могут взаимно превращаться друг в друга, например, в результате реакции таутометрии, поворотной изомерии, ионизации и т.д. Анализ таких смесей представляет одну из наиболее важных задач спектрофотометрии.
Спектрофотометрический метод может быть применен для анализа смеси только в том случае, если спектры компонентов заметно отличаются. Предположим, что спектры чистых веществ А и В имеют вид, показанный на рис. 3.
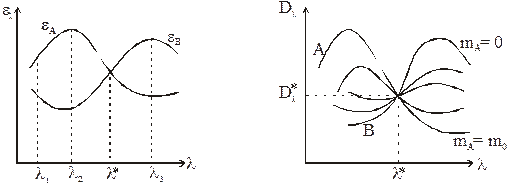
Рис. 3 Рис. 4
Точка пересечения спектральных кривых называется изобестической. На рис. 3 эта точка имеет абсциссу l*. Покажем, что в изобестической точке (при длине волны l*) оптическая плотность раствора, подчиняющегося условию (10), не зависит от состава раствора. По определению
Dl = L(eAmA + eВmВ) (11)
Исключая из уравнений (11) и (10) концентрацию mB, имеем
Dl = LeВm0 + L(eA – eВ)mA (12)
Но в изобестической точке eА
= eВ,
следовательно, ![]() = LeВm0. Таким
образом,
= LeВm0. Таким
образом, ![]() не зависит от состава
раствора.
не зависит от состава
раствора.
Это означает также, что в изобестической точке пересекаются спектры растворов любого состава, подчиняющегося соотношению (10). Этот случай показан на рис. 4. Здесь А и В – спектры чистых компонентов, между ними лежат спектры растворов при 0 < mA < m0. Некоторые системы могут иметь несколько изобестических точек.
Наличие в спектрах изобестической точки облегчает анализ смесей. Если спектральная кривая одного из рабочих растворов не проходит через такую точку, то это может указывать либо на неправильное приготовление растворов (mA + mB № 0), либо на то, что при изменении состава среды изменяются значения экстинкций eA и eВ (нарушение закона Бера).
Выбор характеристической длины волны, на которой проводится анализ, производят исходя из следующих соображений.
Во-первых, точность анализа смеси тем выше, чем больше
значение производной 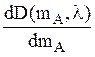 . Согласно (12)
. Согласно (12)
= L (eA – eВ).
(13)
Следовательно, при заданной толщине кюветы L чувствительность растет с увеличением разницы |eA – eВ|, достигая в некоторых точках максимального значения. На рис. 3 это длины волн l2 и l3.
Кроме того, не рекомендуется работать на крутом спаде спектральной кривой, например, в точке l1 (рис. 3), поскольку в этом случае небольшие отклонения в значениях l, связанные, например, с неточностью установки длины волны, могут привести к заметной ошибке в значении Dl и неправильному определению.
Исходя из этого, в качестве характеристической длины волны выбирают l2 или l3, и анализ смесей проводят в этих точках спектра.
Согласно закону Бера отношение D/L должно быть постоянной величиной для данного вещества. Однако в силу конструктивных особенностей спектрофотометров и фотоэлектроколориметров постоянство D/L наблюдается только в среднем интервале оптических плотностей, примерно при 0,1 < D < 1,2 – 1,4. Следовательно, измерения нужно проводить в этой области величин D. Из этого условия подбирают кювету и концентрацию раствора.
Уже отмечалось, что работу начинают с проверки закона Бера. С этой целью исследуют растворы при различных концентрациях m. График D = D(m) должен изображаться прямой, проходящей через нуль (рис. 5).
|
|
1.7. Определение степени диссоциации слабой кислоты
по спектрофотометрическим данным
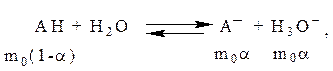
Анализ кислотно-основных равновесий представляет одну из наиболее важных областей приложения спектрофотометрии и колориметрии.
Вода и водные растворы некоторых сильных минеральных кислот и щелочей не поглощают свет в видимой и УФ-областях спектра. Это обстоятельство значительно упрощает применение спектрофотометрии к анализу кислотно-основных равновесий.
 |
(14)
Уважаемый посетитель!
Чтобы распечатать файл, скачайте его (в формате Word).
Ссылка на скачивание - внизу страницы.
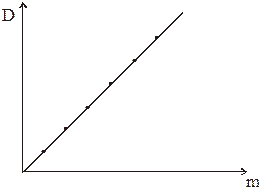 Рис. 5
Рис. 5