


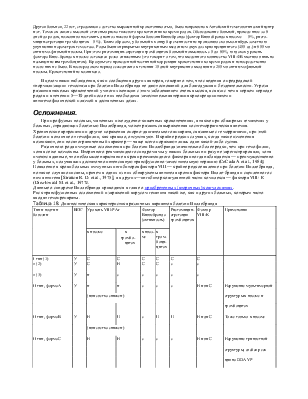



1)гемофилия А - 68-78% 2)болезнь Виллебранда - 9-18% 3)гемофилия В - 6-13%
Это геморрагический диатез, обусловленный наследственным дефицитом или наследственной молекулярной аномалией прокоагулянтной части ф.VIII.
Ф. VIII включает субъединицу VIII:К (носитель коагуляционных свойств, синтезируется в лимфоцитах?, контролируется геном Х-хромосомы) + субъединицу VIII:ФВ или фактор Виллебранда (носитель агрегационной активности Тр, синтезируется в эндотелии, содержится в a-гранулах Тр, контролируется аутосомным геном) + антигенные маркеры.
1)- антигенпозитивная (90-92% больных) - антигеннегативная 2) по уровню фактора: а)крайне тяжелая - 0-1% б)тяжелая - 1-2 % в)средней тяжести - 2-5 % г)легкая - более 5% (возможны тяжелые кровотечения при травмах и хирургических операциях) (60-200 % - нормальный уровень)
- кровоизлияния в крупные суставы конечностей (коленные, локтевые, голеностопные), глубокие подкожные, межмышечные и внутримышечные гематомы, обильные и длительные кровотечения при травмах - возрастная эволюция симптомов: часто после рождения существенных геморрагических проявлений нет (м.б долго незаживающая ранка пупка, кефалогематома), з-ние распознается на 2-3 году; в раннем детском возрасте часты кровотечения из слизистой рта, ягодичной области; затем на первый план выступают геморрагии в крупные суставы - различают а) острые гемартрозы - первичные и рецидивирующие, б) хронические геморрагиччески-деструктивные остеоартрозы, в) вторичный ревматоидный синдром. С возрастом тяжесть и распространенность поражения неуклонно прогрессирует. - гематомы могут быть причиной сепсиса или некроза окружающих тканей, м.б стенозирование верхних дыхательных путей и асфиксия (в диагностике помогаю УЗИ, радионуклидное сканирование с меченными Эр) - почечные крововотечения (у 14-30%) часто сопровождаются дизурией и приступами почечной колики
- возможны ингибиторные формы (высокие титры в крови иммунных ингибиторов ф. VIII) - вторичный ревматоидный синдром - большая эозинофилия - амилоидоз почек - велика вероятность заразиться гепатитом, ВИЧ
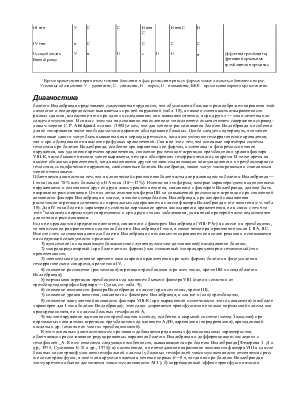
- гипокоагуляция в пробах: парциальное тромбопластиновое время с кефалином и АКТ - показатели тромбинового и протромбинового времени нормальные - при тяжелой форме: удлинение общего времени свёртывания плазмы, снижение потребления протромбина - установка вида гемофилии проводится с помощью коррекционных проб в одном из тестов: 1) тест генерации тромбопластина по Биггс-Дугласу-Макфарлану, 2) тест образования тромбина, 3) антикоагуляционный тест на 4-ой мин инкубации - количественное определение дефицитного фактора
ВНУТРИМЫШЕЧНЫЕ и ПОДКОЖНЫЕ ИНЪЕКЦИИ ПРОТИВОПАКАЗАНЫ!
1) Заместительная терапия. NB! Ф. VIII практически не сохраняется в консервированной крови , нативной и сухой плазме. Период полужизни фактора- 8-12 ч. А)криопреципитат и концентраты фактора 8 (наиболее эффективны) б)антигемофильная плазма (в 3-4 раза эффективнее трансфузии свежей крови) условия: - все препараты должны вводится в/в только струйно в концентрированном виде и возможно быстрее после их расконсервирования - до стойкой остановки кровотечения следует избегать введения любых кровезаменителей и гемопрепаратов. (возможно применение эритромассы) 2)при гемартрозах:
- иммобилизация на 3-5 дней конечности в физиологическом положении, обогревание сустава, ранняя аспирация крови при обширных гемартрозах 3)наружные кровотечения: + местное воздействие тромбопластином, тромбином и охлажденной 5-6% e-АКК - при носовых кровотечениях следует избегать тугой тампонады 4)курсы преднизолона 1-2 мг/кг при почечных кровотечениях и ревматоидном синдроме 5)при ингибиторной форме: концентраты ф.VIII или протромбинового комплекса с трансфузиями тромбоцитарной массы на фоне преднизолона 6)лечебный плазмаферез
Наследственное заболевание, характеризующееся повышенной кровоточивостью в сочетании с увеличением длительности кровотечения, низким уровнем VIII фактора в крови и очень низкими (или отсутствием) адгезии тромбоцитов к стеклу, агрегации тромбоцитов с ристоцетином.
Помимо перечисленных основных типов, в литературе описано и продолжает описываться множество других вариантов нарушения продукции и супермолекулярных аномалий фактора Виллебранда и связанного с ним антигена, в частности, с разным содержанием этих компонентов в эндотелии, плазме и тромбоцитах. Из них особый интерес представляет форма, описанная С. Korninger и соавт. (1981), при которой снижение продукции и высвобождение из эндотелия ФВ (в ответ на компрессию сосудов или введение аргинин-вазопрессина) сочеталось с выраженным нарушением фибринолиза из-за пареза продукции в эндотелии тканевого активатора фибринолиза. Аналогичную форму описали и V. Vicente и соавт. (1984). При этой форме может выявляться наклонность к тромбозам.
Как следует из приведенных данных, число разновидностей болезни Виллебранда огромно и выявление всех ее вариантов еще далеко не знакончено. Это разнообразие определяется, с одной стороны, сложностью надмолекулярной структуры комплекса фактора VIII и изменчивостью взаимодействия входящих в его состав разных активностей, а с другой — участием субъединиц фактора VIII во всех звеньях системы гемостаза (сосудистом, тромбоцитарном и коагуляционном), неоднородностью нарушений в каждом из этих звеньев.
Уважаемый посетитель!
Чтобы распечатать файл, скачайте его (в формате Word).
Ссылка на скачивание - внизу страницы.