Тирозинемия типа II (синдром Рихнера— Ханхарта) развивается при недостаточность первого фермента тирозин-трансаминазы печени. Для клинические проявлений характерны поражения глаз и кожи и умеренная умственная отсталость. Содержания тирозина в плазме повышено (4—5 мг/100 мл), Тирозин является единственной аминокислотой, концентрация которой в моче повышается Среди выделяемых метаболитов выделяемых мочой можно найти пара гидроксифенилпируват, п-гидроксифениллактат, и-гидроксифенилацетат, N-ацетилтирозин и тирамин .
Недостаточность еще одного фермента обмена тирозина пара-гидроксифенил-пируват-гидроксилазы вызывает тирозинемию новорожденных . При этом повышается содержание в крови тирозина и фенилаланина, а в моче содержание тирозина, и-гидроксифенилацетата, N-ацетилтирозина и тирамина. Для лечения назначают бедную белком диету.Нарушение активности оксидазы гомогентизиновой кислоты приводит к развитию алкаптонурии. Это наследственное нарушение метаболизма описано в медицинской литературе еще в XVI в и стало первым заболеванием в истории медицины побудившее Гаррода выдвинуть идею о наследственных метаболических нарушениях. Моча таких больных темнеет при контакте с воздухом. Со временем у человека наблюдается общая пигментация соединительной ткани (охроноз) и развивается артрит. Субстрат фермента, гомогентизат, экскретируется с мочой; при окислении на воздухе он образует темно-коричневый пигмент. Алкаптонурия наследуется по аутосомно-рецессивному типу. В настоящее время не найдено диагностических методов выявления гетерозигот. Хотя механизм охроноза не установлен, полагают, что он обусловлен окислением гомогентизата полифенолоксидазой, приводящим к образованию бензохиноацетата, который далее полимеризуется и связывается с макромолекулами соединительной ткани.
Триптофан одна из первых аминокислот отнесенных к незаменимым. Основная масса поступившего с пищей триптофана используется в синтезе белков. Около 1% расходуется на образование серотонина и мелатонина. Распад молекулы триптофана до промежуточных продуктов проходит в основном по кинуренин-антранилатному пути. Триптофаноксигеназа -первый фермент этого пути представляет металлопротеин, содержащий железопорфириновый комплекс. Этот фермент катализирует раскрытие индольного кольца с включением двух атомов молекулярного кислорода и образованием N-формилкинуренина. Повышение уровня триптофана вызывает индукцию синтеза этого фермента, триптофан повышает его стабильность в клетке. Фермент ингибируется по принципу обратной связи производными никотиновой кислоты. Кинуренин-формилаза –второй фермент процесса катализирует гидролитическое удаление формильной группы N-формилкинуренина и и образование кинуренина. Последний превращается в 3-гидроксикинуренин и затем в 3-гидроксиантраниловую кислоту. Гидроксилирование происходит аналогично реакции гидроксилирования фенилаланина при участии молекулярного кислорода. Кинурениназа , катализирующая образование гидроксиантраниловой кислоты пиридоксальфосфат зависимый фермент. Эта реакция чувствительна к недостатку витамина В6. При его недостаточности происходит нарушение образования гидроксиантраниловой кислоты и производные кинуренина во внепеченочных тканях превращаются в ксантуреновую кислоту. Ксантуреновая кислота при достаточном поступлении витамина В6 в моче не обнаруживается.
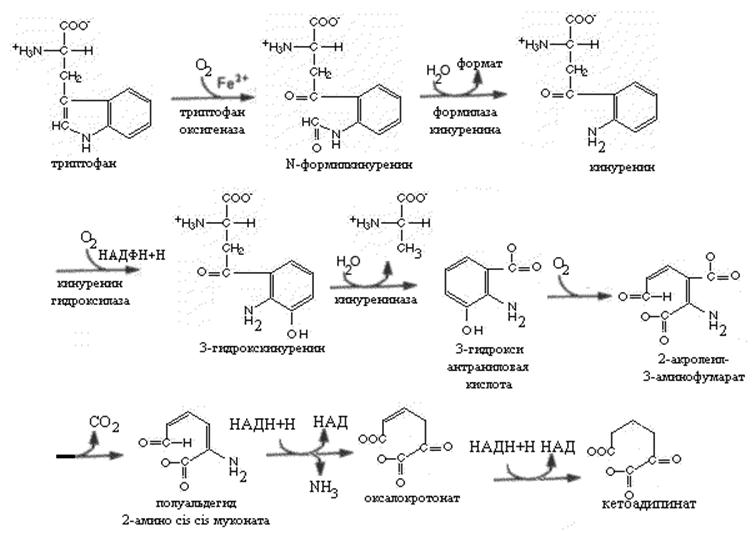
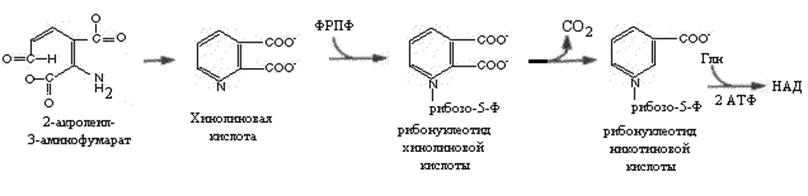
У многих животных часть 2-акролеил-3-аминофумарата превращается в хинолиновую кислоту, из которой после ряда реакций с участием ФРПФ, глутамина и 2 молей АТФ образуется НАД. У многих животных превращение триптофана в никотиновую кислоту делает необязательным поступление этого витамина с пищей (крысы, кролики, собаки свиньи).Введение человеку избыточного количества триптофана с пищей повышает экскрецию с мочой производных никотиновой кислоты (например, N-метилникотинамида).
 |
Врожденное нарушение обмена триптофана, проявляющееся появлением сыпи на коже, перемежающейся мозжечковой атаксией и умственной отсталостью известно под названием болезни Хартнупа, В моче таких больных обнаруживаются повышенные количества индолацетата и триптофана.
Треонин и лизин - незаменимые у млекопитающих аминокислоты. Они отличаются от остальных аминокислот тем, что при своем распаде они не подвергаются дезаминированию и следовательно не образуют соответствующих кетокислот, из которых их можно было бы получить. У микроорганизмов синтез треонина и лизина начинается преобразованием оксалоацетата в аспарагиновый полуальдегид. Этот путь использует одну молекулу АТФ и две молекулы НАДФH + Н+
Биосинтез треонина завершается в три этапа. Вначале реакция восстановления при участии НАДФH + Н + дает гомосерин, который после фосфорилирования в гомосеринфосфат с затратой АТФ превращается в треонин
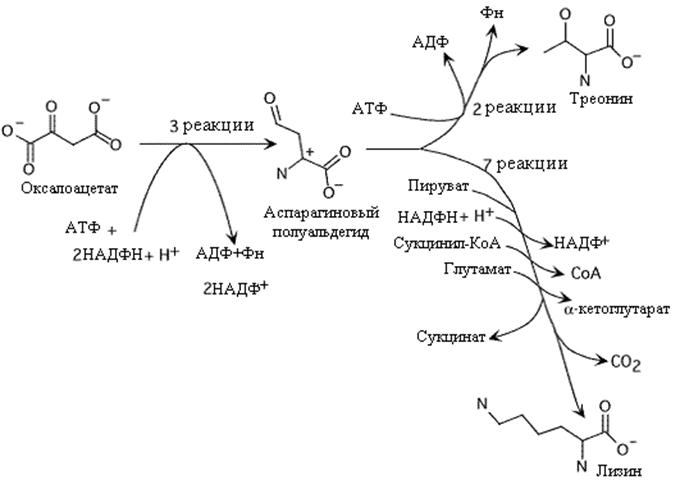
. Синтез треонина и лизина. Обратите внимание на количество энергии, которая израсходована в этих биосинтезах.
На рис приводится обобщенный путь синтеза лизина, хотя он различается у разных бактерий. Синтез лизина включает присоединение пирувата к аспарагиновому полуальдегиду, использование КоА производных (или ацетил-КоА или сукцинил-КoA) и присединение аминогруппы глутамата. Ацил-КоА (сукцинил, или ацетил) играют роль блокирующей группы, защищающей аминогруппу от атаки во время переаминирования глутаматом. NAДФH +Н+ используется для восстановления на втором этапе пути.
Уважаемый посетитель!
Чтобы распечатать файл, скачайте его (в формате Word).
Ссылка на скачивание - внизу страницы.