Гнперфеннлаланинемия типа I (классическая фенилкетонурия, ФКУ) проявляется умственной отсталостью, припадками, психозами, экземой и появлением «мышиного» запаха. Указанные симптомы при раннем диагнозе и своевременном лечении могут не проявиться. Классическая ФКУ встречается с частотой 1 на 10000 новорожденных, активность фермента составляет до 25% от нормы и фермент утрачивает чувствительность к регуляторному действию фенилаланина.
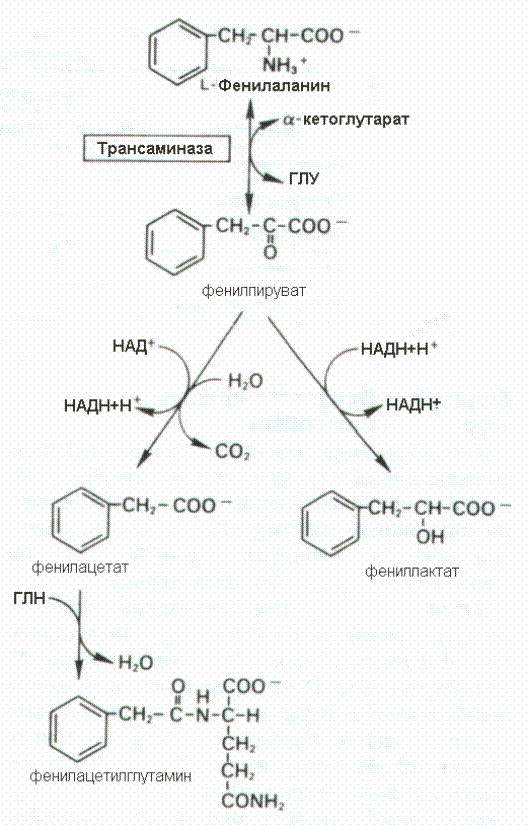
Нарушение превращения в тирозин резко усиливает альтернативные пути превращения фенилаланина. В крови увеличивается уровень фенилаланина, растет его выделение почками, а в крови и моче появляются фенилпируват, фениллактат, фенилацетат и фенилацетилглутамин. Появление в крови и моче кетокислоты фенилпирувата определило и само название болезни—фенилкетонурия. Для выявления этого заболевания помимо открытия присутствия фенилпирувата в моче необходимо определить содержание фенилаланина в плазме крови. Ранняя диагностика и назначение диеты с очень низким содержанием фенилаланина позволяет предотвратить все симптомы и неблагоприятные последствия этого заболевания. Эту диету следует использовать до достижения шестилетнего возраста, когда снижается неблагоприятное влияние на развитие мозга промежуточных метаболитов фенилаланина.
Фенилаланин может подвергаться переаминированию с a-кетоглутаровой кислотой с образованием фенилпирувата. Фенилпировинорадная кислота восстанавливается с образованием фениллактата или подвергается окислительному декарбоксилированию, продукт которого связываясь с глутамином в форме фенилацетилглутамина выделяется. Эти реакции протекает в печени у здоровых людей, однако количество катаболизируемого таким образом фенилаланина незначительно. Эти пути приобретают значимость при нарушении функции фенилаланингидроксилазы
 |
Нарушение функции фенилаланин гидроксилазы – причина фенилкетонурии
Учитывая указанные выше особенности функции фенилаланингидрокислазы, могут быть три основные причины нарушения превращения фенилаланина в тирозин
катаболизируемого таким образом фенилаланина незначительно. Эти пути приобретают значимость при нарушении функции фенилаланингидроксилазы
|
Нарушение функции фенилаланин гидроксилазы – причина фенилкетонурии
Учитывая указанные выше особенности функции фенилаланингидрокислазы, могут быть три основные причины нарушения превращения фенилаланина в тирозин
Гнперфеннлаланинемия типа I (классическая фенилкетонурия, ФКУ) проявляется умственной отсталостью, припадками, психозами, экземой и появлением «мышиного» запаха. Указанные симптомы при раннем диагнозе и своевременном лечении могут не проявиться. Классическая ФКУ встречается с частотой 1 на 10000 новорожденных, активность фермента составляет до 25% от нормы и фермент утрачивает чувствительность к регуляторному действию фенилаланина.
Нарушение превращения в тирозин резко усиливает альтернативные пути превращения фенилаланина. В крови увеличивается уровень фенилаланина, растет его выделение почками, а в крови и моче появляются фенилпируват, фениллактат, фенилацетат и фенилацетилглутамин. Появление в крови и моче кетокислоты фенилпирувата определило и само название болезни—фенилкетонурия. Для выявления этого заболевания помимо открытия присутствия фенилпирувата в моче необходимо определить содержание фенилаланина в плазме крови. Ранняя диагностика и назначение диеты с очень низким содержанием фенилаланина позволяет предотвратить все симптомы и неблагоприятные последствия этого заболевания. Эту диету следует использовать до достижения шестилетнего возраста, когда снижается неблагоприятное влияние на развитие мозга промежуточных метаболитов фенилаланина.
При изучении основных путей катаболизма тирозина важную роль сыграли исследования генетических заболеваний. Первая реакция катаболизма тирозина подобна таковой для фенилаланина – трансаминирование с образованием парагидроксифенил-пирувата, которое катализируется тирозин-a-кетоглутарат-трансаминазой в печени млекопитающих.
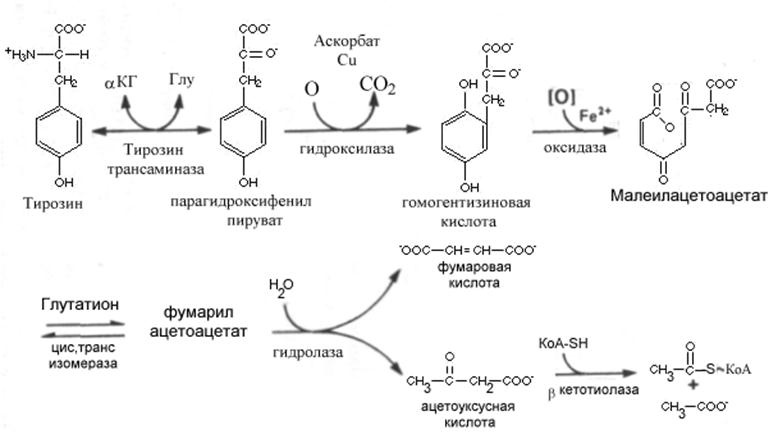
Превращение в гомогентизиновую кислоту происходит при участии пара гидроксифенилпируват гидроксилазы. Этот медь содержащий фермент катализирует сложную реакцию, сопровождающуюся переносом боковой цепи, декарбоксилированием и гидроксилированием кольца. Кофактором этой реакции, по-видимому, служит аскорбиновая кислота, поскольку больные цингой выделяют мочой промежуточные продукты метаболизма тирозина. Раскрытие кольца гомогентизиновой кислоты происходит под влиянием оксидазы гомогентизиновой кислоты.— железосодержащего металлопротеина печени млекопитающих. Образующийся малеилацетоацетат превращается в свой изомер фумарилацетоацетат при участии цис, трансизомеразы и гидролаза фумарилацетоацетата завершает катаболический путь тирозина образованием фумарата и ацетоацетата, последний может превратиться в ацетил-КоА.
При недостаточности ферментов катализирующих заключительные этапы катаболизма тирозина развивается тирозиноз или тирозинемия типа I. При этомнакапливаются промежуточные продукты, оказывающие ингибирующее действие на ряд ферментов и транспортных систем с недостаточно изученным механизмом действия. Различают острую и хроническую формы тирозинозов. Острая форма развивается в раннем детстве и проявляется в форме нарушений деятельности желудочно-кишечного тракта (понос, рвота) «капустного» запаха, задержки в развитии. Недостаточность печени и смертельный исход наступает в возрасте 6—8 месяцев при несвоевременном лечении. Со сходными, но более умеренно выраженными симптомами, протекает хроническая тирозинемия. Летальный исход наступает в возрасте около 10 лет. Содержание тирозина в плазме повышается до 6—12 мг/100 мл, повышено содержание и некоторых других аминокислот, особенно метионина. Лечение включает диету с пониженным содержанием тирозина и фенилаланина, а в ряде случаев также и метионина.
Уважаемый посетитель!
Чтобы распечатать файл, скачайте его (в формате Word).
Ссылка на скачивание - внизу страницы.