К нейрогуморальным механизмам компенсации относятся активация ренин-ангиотензин-альдостероновой (тканевой и циркулирующей) и симпатоадреналовой систем. Ренин синтезируется преимущественно почками. Он превращает ангиотензиноген, образующийся главным образом в печени, в ангиотензин I. Ренин секретируется в ответ на снижение артериального давления, уменьшение содержания натрия в крови и симпатическую стимуляцию почек, а также диуретическую терапию. Образовавшийся ангиотензин I превращается затем под влиянием ангиотензинпревращающего фермента и хемаз (в основном в лёгких) в ангиотензин II, который является мощным прессорным агентом, способствует гипертрофии миоцитов сердечно-сосудистой системы, ускоряет запрограммированную гибель клеток миокарда (апоптоз), процессы фиброза сердечной мышцы и ухудшает диастолическое расслабление. Всё это отражает важную роль ангиотензина II в патологическом ремоделировании сердца при СН. Под влиянием этого вещества происходит увеличение синтеза альдостерона в надпочечниках, который в свою очередь задерживает натрий и воду, увеличивает экскрецию калия, стимулирует процессы фиброза в сосудистой стенке и миокарде. Разрушение этого гормона происходит в печени, поэтому при наличии застойных явлений в печени или снижении печёночного кровотока уровень альдостерона может длительное время оставаться повышенным, обеспечивая его эффекты. Под воздействием ангиотензина II увеличивается секреция в ядрах гипоталамуса антидиуретического гормона, который способствует задержке жидкости, что в совокупности с другими факторами обуславливает возникновение отёчного синдрома. Кроме ренин-ангиотензин-альдостероновой системы, в процессах компенсации большую роль играет симпатоадреналовая система, активность которой резко повышается при активации барорецепторов синокаротидной зоны и дуги аорты в ответ на снижение артериального давления в большом круге кровообращения, активации вазомоторного центра в связи с гипоксией мозга, а также рефлекса Бейнбриджа с растягивающихся устьев полых вен. При этом снижается тонус вагуса и возрастает тонус симпатической нервной системы. По мере прогрессирования СН снижается чувствительность барорецепторов, что приводит к нарушению регуляторного механизма отрицательной обратной связи. В целом, активация этих систем приводит на ранних этапах к поддержанию уровня артериального давления за счёт увеличения общего периферического сосудистого сопротивления и объёма циркулирующей плазмы, а также к увеличению наполнения полостей сердца в связи с констрикцией венозного русла и увеличения венозного возврата, а также к снижению способности к вазодилатации различных сосудистых сетей, в связи с утолщением мышечного слоя сосудов. Но увеличение общего периферического сосудистого сопротивления приводит к увеличению преднагрузки на сердце, а увеличения венозного возврата представляет повышение постнагрузки. Оба этих процесса приводят к увеличению работы сердца, ускоряя наступление декомпенсации. Кроме того, гиперкатехоламинемия влечёт за собой спазм сосудов почек, существенно нарушая их функцию, приводя к снижению диуреза и накоплению жидкости, а также вызывает тахикардию, которая с одной стороны способствует поддержанию минутного объёма кровообращения, а с другой стороны истощает энергетические запасы кардиомиоцитов, увеличивает работу сердца и сокращает диастолу, ухудшая процесс наполнения камер сердца и его питания. К тому же, катехоламины увеличивают электрическую возбудимость миокарда, повышая риск возникновения нарушений ритма, которые в свою очередь затрудняют обеспечение нормальной гемодинамики, а также приводят к увеличению агрегационной активности тромбоцитов, ухудшая реологические свойства крови и повышая риск тромбообразования.
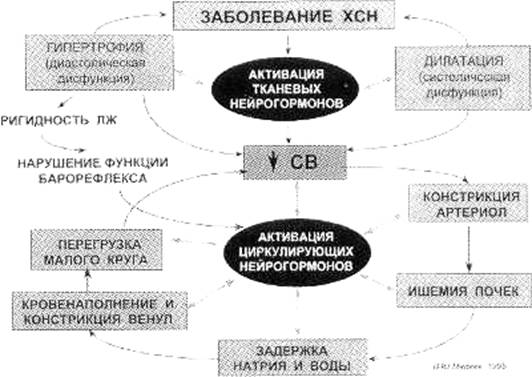 Увеличение конечного
диастолического давления в левом предсердии является пусковым фактором для
продукции натрийуретического фактора. Последний оказывает антагонистическое
воздействие на ренин-ангиотензин-альдостероновую систему и в какой-то степени
защищает сосудистую систему от избыточной вазоконстрикции, способствуя снижению
секреции ренина, альдостерона, вазопрессина, увеличивая медуллярный кроваток в
почках и приводя к снижению ретенции жидкости в организме. Но по мере
прогрессирования СН снижается способность поражённого сердца синтезировать этот
фактор, таким образом, уменьшаются и его протекторные эффекты на сосудистое
русло. При СН увеличивается секреция эндотелием местного вазоконстриктора – эндотелина,который помимо увеличения общего
периферического сосудистого сопротивления совместно с вышеуказанными системами
участвует в так называемой централизации кровообращения. Это механизм
поддерживает достаточный кроваток в жизненно важных органах и приводит к
резкому ограничению кровоснабжения периферических тканей, вызывая трофические
изменения кожи, мышечные атрофии и хронические ишемические и дистрофические
изменения в менее важных внутренних органах. Кроме того, этот пептид принимает
участие в увеличении артериального давления в лёгочном круге кровообращения,
что сопровождается перегрузкой правых отделов сердца и приводит к их
недостаточности.
Увеличение конечного
диастолического давления в левом предсердии является пусковым фактором для
продукции натрийуретического фактора. Последний оказывает антагонистическое
воздействие на ренин-ангиотензин-альдостероновую систему и в какой-то степени
защищает сосудистую систему от избыточной вазоконстрикции, способствуя снижению
секреции ренина, альдостерона, вазопрессина, увеличивая медуллярный кроваток в
почках и приводя к снижению ретенции жидкости в организме. Но по мере
прогрессирования СН снижается способность поражённого сердца синтезировать этот
фактор, таким образом, уменьшаются и его протекторные эффекты на сосудистое
русло. При СН увеличивается секреция эндотелием местного вазоконстриктора – эндотелина,который помимо увеличения общего
периферического сосудистого сопротивления совместно с вышеуказанными системами
участвует в так называемой централизации кровообращения. Это механизм
поддерживает достаточный кроваток в жизненно важных органах и приводит к
резкому ограничению кровоснабжения периферических тканей, вызывая трофические
изменения кожи, мышечные атрофии и хронические ишемические и дистрофические
изменения в менее важных внутренних органах. Кроме того, этот пептид принимает
участие в увеличении артериального давления в лёгочном круге кровообращения,
что сопровождается перегрузкой правых отделов сердца и приводит к их
недостаточности.
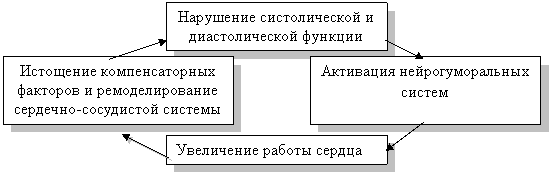
Таким образом, в результате ремоделирования сердечно-сосудистой системы и постепенного истощения компенсаторных факторов сердце вступает в фазу «прогрессирующего кардиосклероза и изнашивания структур, которая характеризуется нарушением обновления структур и влечёт за собой гибель клеток, развитие склероза органа. Прогрессирующая гипертрофия и дилатация сердца сопровождаются дальнейшим нарушением систолической и диастолической функций желудочков, увеличением потребности миокарда в кислороде, нарушениями биоэнергетики миокарда, изменением субэндокардиального кроватока, и увеличением риска развития опасных для жизни желудочковых аритмий.
Уважаемый посетитель!
Чтобы распечатать файл, скачайте его (в формате Word).
Ссылка на скачивание - внизу страницы.