Выделяют исследования при
низком ![]() Å), среднем (3,5 Å > >
Å), среднем (3,5 Å > > ![]() > 2,5 Å) и высоком
> 2,5 Å) и высоком ![]() Å) разрешениях. По картам
электронной плотности низкого разрешения, как правило, не удается однозначно
выявлять ход полипептидной цепи. Однако трехмерные модели электронной
плотности, построенные на основе таких данных, часто дают правильное
представление об общей конфигурации молекулы белка. Карты электронной плотности
среднего разрешения обычно позволяют проследить ход полипептидной цепи, однако
их интерпретация зависит от предварительного знания стереохимии аминокислотных
остатков. В случае карт электронной плотности высокого разрешения
обнаруживается дальнейшее улучшение детальности картины. Например, при dm=1,9 Å на карте электронной плотности
белка инсулина разрешаются атомы серы дисульфидной связи, уточняется
конформация полипептидного остова, лучше выявляются ароматические остатки.
Å) разрешениях. По картам
электронной плотности низкого разрешения, как правило, не удается однозначно
выявлять ход полипептидной цепи. Однако трехмерные модели электронной
плотности, построенные на основе таких данных, часто дают правильное
представление об общей конфигурации молекулы белка. Карты электронной плотности
среднего разрешения обычно позволяют проследить ход полипептидной цепи, однако
их интерпретация зависит от предварительного знания стереохимии аминокислотных
остатков. В случае карт электронной плотности высокого разрешения
обнаруживается дальнейшее улучшение детальности картины. Например, при dm=1,9 Å на карте электронной плотности
белка инсулина разрешаются атомы серы дисульфидной связи, уточняется
конформация полипептидного остова, лучше выявляются ароматические остатки.
Значительно ускорить интерпретацию карт электронной плотности среднего и высокого разрешения позволяет изготовление моделей. Сопоставление модели с электронной плотностью позволяет снизить возможность ошибок при интерпретации электронной плотности. Наиболее универсальным способом такого сопоставления модели белка с электронной плотностью является применение машинной графики. В результате получают окончательную модель белка, которая часто представляет последнюю стадию рентгеноструктурного анализа. Такие модели характеризуют вторичную и третичную структуры белка и, соответственно, существенно развивают представление о биологических молекулах, к которым относятся ферменты, белковые гормоны, транспортные белки, иммуноглобулины.
Однако эти модели далеки от совершенства. Поэтому кристаллографы, исследующие белки, обязаны улучшать их. Основными недостатками, характерными для таких результатов рентгеноструктурного анализа, являются:
а) неточность фаз, которые были определены, как описано выше, связанная как со случайными погрешностями, обусловленными неточностью измерений, так и с погрешностями из-за нарушения изоморфизма кристаллов нативного белка и производных;
б) невозможность в некоторых случаях определения фаз отражений для межплоскостных расстояний d<2 Å при использовании нескольких изоморфных производных (хотя рефлексы на рентгенограмме нативного белка можно наблюдать и при более высоком разрешении);
в) не лучшее соответствие изготовленной модели рассчитанной электронной плотности.
Поэтому для уточнения модели белка можно уточнить фазы, расширить их на область более высокого разрешения, а также строить модель таким образом, чтобы она была наилучшим отображением рассчитанной электронной плотности.
Есть различные методики уточнения модели белка, и с ними можно ознакомиться в цитируемой литературе. Отметим, что практически во всех успешных уточнениях структур белков на одной из стадий анализа использовались разностные синтезы Фурье, которые позволяют, как утверждается, особенно просто обнаруживать грубые ошибки.
Разностные синтезы Фурье
также используются для изучения соединений белковых молекул с малыми
молекулами. Впервые этот метод был применен для изучения связывания миоглобином
азид-иона. В дальнейшем он широко применялся для исследования связывания
ингибиторов и псевдосубстратов многими белками. Этот метод дает возможность
локализации активных центров ферментов. Разностные синтезы Фурье позволяют
фиксировать конформационные изменения в структуре нативного белка,
соответствующие сдвигам атомов, не превышающим 1 Å. Ранее мы уже рассматривали
применение этих синтезов для определения координат тяжелых атомов. Выражения
для коэффициентов в этом случае подобны приведенному ранее (2.21), только
вместо ![]() используется модуль структурного
фактора белка с ингибитором
используется модуль структурного
фактора белка с ингибитором ![]() . Потому
разностный синтез Фурье
. Потому
разностный синтез Фурье ![]() имеет вид
имеет вид
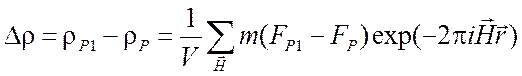 ,
(2.36)
,
(2.36)
где ![]() и
и ![]() –
электронные плотности нативного белка и белка с ингибитором соответственно, а
суммирование проводится по всем отражениям. Разность
–
электронные плотности нативного белка и белка с ингибитором соответственно, а
суммирование проводится по всем отражениям. Разность ![]() является электронной плотностью
ингибитора.
является электронной плотностью
ингибитора.
И, наконец, отметим, что преимущество разностных синтезов Фурье состоит также в отсутствии ошибок обрыва ряда.
Первым белком, пространственная
структура которого была установлена с помощью рентгеноструктурного анализа, был
миоглобин (группа Дж.Кендрью, 1957 г., Кембридж, Англия). Молекула миоглобина
имеет массу около 17000, содержит в единой полипептидной цепи 153
аминокислотных остатка и группу гемма, в которую входит атом железа. Вначале
структура миоглобина была получена с довольно низким разрешением (6 Å), для
чего потребовалась учитывать в синтезе Фурье 400 рефлексов (рис. 2.16,а). В
результате полученная структура показывала лишь контуры спиральных участков молекулы.
Через 2 года эта же группа получила пространственную конфигурацию миоглобина с
разрешением 2 Å (учитывались 9600 дифракционных рефлексов). Несмотря на то, что
отдельные атомы в такой структуре не видны, удалось локализовать ![]() атомы полипептидной цепи, а также
атомы боковых радикалов. В дальнейшем пространственная структура миоглобина
была определена с еще большим разрешением. В настоящее время структура этой
молекулы расшифрована с так называемым атомным разрешением, т.е. разрешением
около 1 Å (рис. 2.16, б). Кроме того, методом рентгеноструктурного анализа
изучены различные функциональные состояния этого белка.
атомы полипептидной цепи, а также
атомы боковых радикалов. В дальнейшем пространственная структура миоглобина
была определена с еще большим разрешением. В настоящее время структура этой
молекулы расшифрована с так называемым атомным разрешением, т.е. разрешением
около 1 Å (рис. 2.16, б). Кроме того, методом рентгеноструктурного анализа
изучены различные функциональные состояния этого белка.
Работы по установлению структур белков после первой успешной расшифровки продолжались и расширялись. К настоящему времени учеными разных стран, среди которых находится и наша страна, исследованы десятки тысяч структур как белков, так и их производных (комплексы с субстратами, ингибиторами и т.п.). В результате накоплен большой статистический материал, хранящийся в Международном банке белковых структур РДВ, адрес Интернет-сайта которого http://www.rcsb.org/rdb/. На данном сайте можно получить доступ к структурам всех белков, изученных методом рентгеноструктурного анализа к настоящему времени.
Существует также программа Ras Mol (http: // www. umas.edu/microbio/rasmol/), с помощью которой осуществляются визуализация и изучение белковых структур, имеющихся в файлах указанного банка.
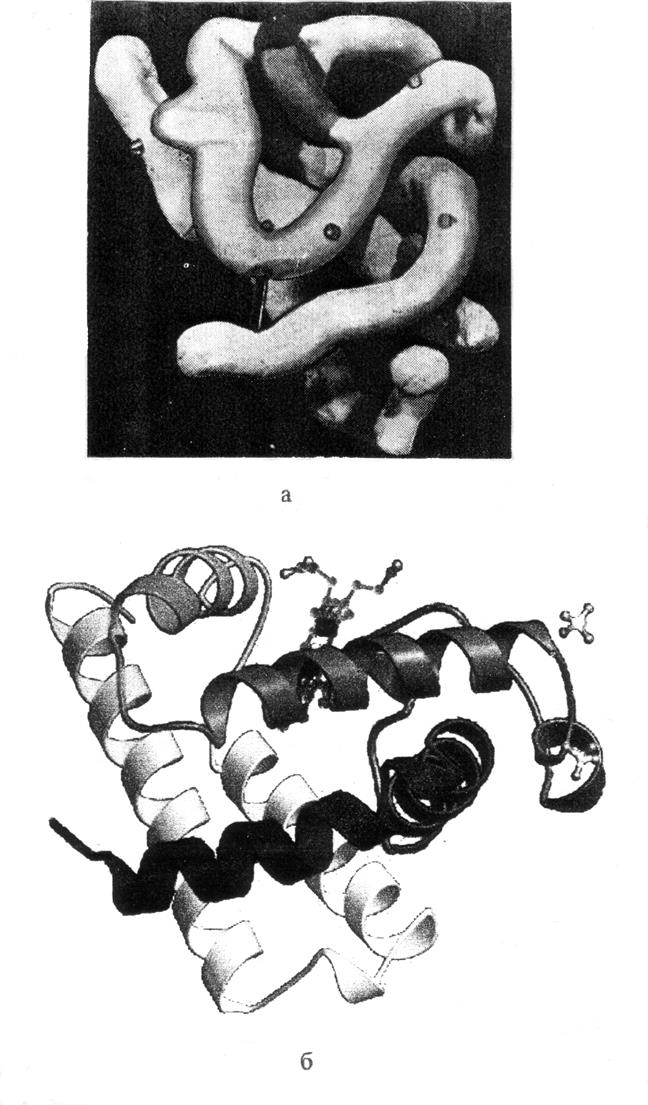
Рис. 2.16. Пространственные структуры молекулы миоглобина,
полученные на основе рентгеноструктурного анализа:
а - ранняя модель молекулы, построенная с разрешением 6 Å;
б - современная модель с атомным разрешением 1 Å, на которой
спиральные участки представлены в виде лент
Уважаемый посетитель!
Чтобы распечатать файл, скачайте его (в формате Word).
Ссылка на скачивание - внизу страницы.