Большая часть спектрометров снабжается несколькими различными изогнутыми кристаллами, каждый из которых может быть установлен в рабочее положение поворотом держателя без нарушения вакуума. Кроме того, поскольку рентгеновские лучи от образца распространяются одновременно в разные стороны, используются по крайней мере два отдельных спектрометра, что позволяет одновременно вести исследование сразу на несколько элементов.
В некоторых конструкциях предусматривается анализ энергетической дисперсии с использованием твердотельных детекторов. Однако энергетическое разрешение этих приборов пока невелико (~250 эВ). Это ограничивает их применимость только анализом элементов в диапазоне от Z = 12 до Z = 92. Их преимуществом является экспрессность анализа, однако при точных измерениях следует предпочесть кристаллические дифрактометры.
Обязательной процедурой при рентгеновском микроанализе является определение рентгеновского излучения эталона, содержащего 100% исследуемого элемента, и фона или шума, главным источником которого в данном случае является непрерывный тормозной спектр.
Для оценки химического состава с точностью до десятых долей процента концентрации элемента Х используется простое выражение:
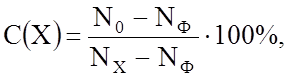 (3.4.1)
(3.4.1)
где N0, NX и NФ - интенсивности рентгеновского излучения данной длины волны l эталона, образца и фона соответственно. Величины N0, NX и NФ регистрируются или путем непосредственной цифровой индикации, или по дифрактограммам в линейных величинах. Очень часто в числителе формулы (3.4.1) NФ можно пренебречь в сравнении с N0.
Точный количественный анализ требует учета поправок на поглощение, так как характеристическое рентгеновское излучение, возбуждаемое на некоторой глубине, частично поглощается по мере прохождения к поверхности. Если атомные номера основного вещества матрицы и легирующей добавки различаются более чем на 4 единицы, требуется еще введение поправки на атомный номер. Часто при сопоставлении образцов с одинаковой матрицей, но различными добавками, используются калибровочные графики, а при анализе сложных образцов - сравнение с эталонными специально подготовленными образцами. В последнее время расчетная часть экспериментов все чаще выполняется с помощью встроенной микроЭВМ.
Для получения удовлетворительных повторяющихся результатов, особенно при количественных измерениях, кроме настройки прибора, необходимо с большой тщательностью готовить образцы. Анализируемая поверхность должна быть совершенно плоской и на ней должны отсутствовать царапины и трещины. Было показано, что канавка глубиной 0,5 мкм при угле выхода рентгеновских лучей 20о может привести к ошибке в концентрации, например, магния, на 10%. Если для оптического выявления структуры нельзя избежать травления поверхности, то следует в районе исследования сделать несколько отпечатков микротвердомера, а затем снова механически сполировать протравленный слой, оставив лишь реперные точки-отпечатки.
Образцы должны обладать тепло- и электропроводностью для предотвращения накопления электрических зарядов и тепла, способных внести значительные искажения в результаты измерений. Если весь образец или какая-либо его часть не являются металлом, следует напылить на всю его поверхность очень тонкую (100...1000Å) угольную пленку, слои алюминия, золота или меди - элементов, определение которых в данном исследовании не предполагается. Эти пленки полупрозрачны и микроструктура поверхности через них видна.
Размеры образцов: диаметр в плоскости шлифа - до 18 мм, высота - не более 7...10 мм.
Рентгеноспектральный микроанализ позволяет определять химический состав образца в интервале концентраций от 0,1 до 100% с относительной погрешностью 2% от измеряемой величины, а в некоторых случаях, при тщательном учете поправок и точном измерении интенсивности, погрешность может быть понижена до 1%. Вообще теоретический предел разрешения метода определяется отношением сигналов “линия/фон” и может достигать 3000/1, что соответствует возможности определять минимальное содержание вещества на уровне 0,03%, однако на практике оказываются достоверными результаты, указываемые с точностью 0,1% для не слишком легких элементов.
В табл. 3.4.1 указана предельная чувствительность рентгеновского микроанализа при определении различных элементов. При содержании, меньшем 0,1%, речь может идти лишь о качественном анализе.
Литий, гелий и водород этим методом вообще не определяются, да и все данные для элементов с Z < 11 не слишком надежны. Особые ограничения здесь - при определении содержания углерода, азота и кислорода, так как в подавляющем большинстве используемых приборов вакуум создается механическими и диффузионными масляными насосами, и избежать попадания паров масла, содержащих углерод, на поверхность образца не представляется возможным. Кислород и азот адсорбируются поверхностью из остаточной атмосферы колонны, хотя частично и могут быть удалены прогревом образца под электронным пучком в колонне.
Таблица 4.1
Чувствительность анализа частиц размером ~1 мкм
|
Предельное количество, поддающееся обнаружению, в % от инородной матрицы |
Определяемый элемент |
|
> 10 |
Z = 4 (Be) |
|
1 - 10 |
От Z = 5 (B) до Z = 9 (F) |
|
0,1 – 1 |
От Z = 11 (Na) до Z = 21 (Se) От Z = 33 (As) до Z = 37 (Rb) |
|
0,01 – 0,1 |
От Z = 22 (Ti) до Z = 32 (Ge) От Z = 38 (Sr) до Z = 74 (W) От Z = 82 (Pb) до Z = 92 (U) |
|
0,01 |
От Z = 75 (Re) до Z = 81 (Tl) |
Еще один недостаток, органически присущий методу в варианте с кристаллом-анализатором - большая длительность определения. Скорость сканирования луча или образца под лучом изменяется в пределах от 1 до 50 мкм/мин и не может быть увеличена, так как детектор должен накопить статистически значимое количество импульсов от каждой исследуемой точки.
На рис. 3.4.4 приведен пример вида информации, полученной на рентгеновском микроанализаторе. Исследовалась устойчивость включения из А12O3 диаметром 50 мкм в никелевой матрице после обработки расплавом СaF2. Видно, что после взаимодействия с расплавом шлака, содержащего CaF2, внешние слои включения обогатились кальцием, произошло частичное растворение включения. В нижней части рисунка показан вид структуры в оптическом микроскопе, горизонтальная линия - траектория электронного зонда, с помощью которого получены представленные концентрационные кривые.
В заключение отметим, что лазерный масс-анализатор, свободный от ограничений по массам анализируемых элементов, не дает выигрыша в локальности в сравнении с рентгеновским микроанализом. В лазерном масс-анализаторе импульсами излучения достигается испарение материала в исследуемой точке поверхности. Содержание легких элементов также может быть определено ионными масс-спектрографами, в которых поверхность образца зондируется фокусированным ионным пучком.
Процесс сопровождается выбиванием ионов образца, которые, в свою очередь, разделяются и фокусируются магнитным и электростатическим полями на фотопленку как в лазерном масс-анализаторе, или на ионизационные детекторы. Локальность ионного масс-анализа также не лучше 2...3 мкм.
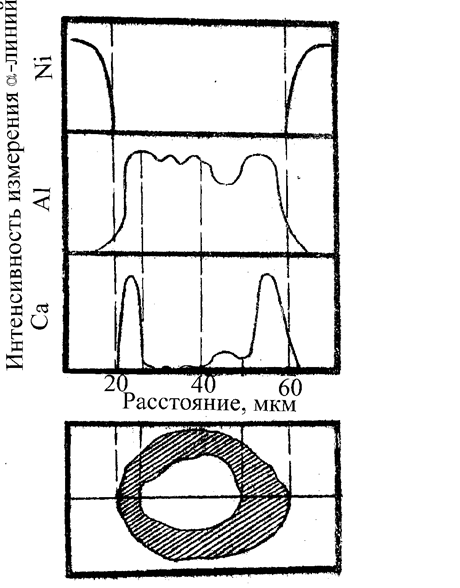 |
Рис. 3.4.4. Распределение Ni, Al, Ca в матрице и включении Al2O3 после погружения в расплав СaF2 на 1,5 мин. На концентрационных кривых отмечается "дополнение" кальцием до 100% содержания концентрации вымываемого алюминия. Граница включения определяется точками падения содержания Ni до нуля
В процессе лазерного и ионного масс-анализа поверхностный слой образца по траектории пучка испаряется, на поверхности остается видимый след воздействия. Микрорентгеноспектральный анализ не разрушает исследуемый материал.
Уважаемый посетитель!
Чтобы распечатать файл, скачайте его (в формате Word).
Ссылка на скачивание - внизу страницы.