13
Рост популярности внутривенной анестезии обусловлен разработкой быстро- и короткодействующих внутривенных гипнотиков, аналгетиков и миорелаксантов, а также надежного, простого в употреблении инфузионного оборудования (табл. 13-1 и рис. 13-1) (Van Hemelrijck J, White PF: Nonopioid intravenous anesthesia. In Barash PG, Gullen BF, Stoelting RK [eds]: Clinical Anesthesia, pp 311-327. Philadelphia, Lippincott-Raven, 1997). Поскольку желаемые фармакологические свойства не одинаково важны в каждой клинической ситуации, анестезиолог должен выбирать анестетик, исходя из индивидуальных особенностей пациента и оперативного вмешательства.
A. Механизм действия
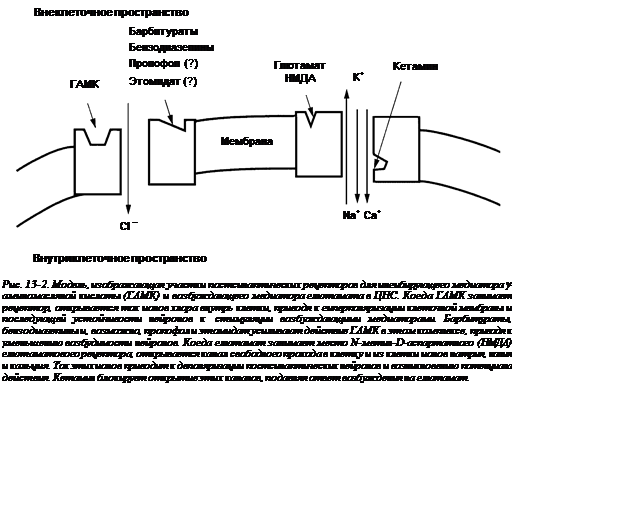
1. Существует признанная теория, согласно которой внутривенные гипнотики (бензодиазепины, барбитураты, пропофол, этомидат, кетамин) оказывают свое влияние, главным образом, через взаимодействие с ингибирующим нейротрансмиттером — g‑аминомасляной кислотой (ГАМК) (рис. 13-2).
2. Активация комплекса «ГАМК-рецептор» увеличивает трансмембранное поступление ионов хлора, приводящее к гиперполяризации и функциональному подавлению постсинаптического нейрона.
3. Бензодиазепины увеличивают эффективность соединения ГАМК с ее рецепторами («потолковый эффект»).
4. Барбитураты и пропофол уменьшают диссоциацию ГАМК и рецепторов.
5. Считается, что основное действие кетамина на ЦНС обусловливается его антагонизмом к N-метил-D-аспартатным (НМДА) рецепторам (аналгетические свойства также могут опосредоваться через стимуляцию опиоидных спинальных рецепторов).
B. Фармакокинетика и метаболизм
1. Быстрое начало действия на ЦНС большинства внутривенных анестетиков может объясняться их высокой жирорастворимостью и относительно высоким уровнем мозгового кровотока.
2. Фармакокинетика внутривенных гипнотиков характеризуется быстрыми распределением и перераспределением в нескольких гипотетических камерах с последующим их выведением (табл. 13-2).
a. Основным механизмом прекращения действия на ЦНС препаратов, используемых для внутривенного вводного наркоза, является перераспределение их из центральной высоко перфузируемой камеры (мозг) в бóльшую и хорошо перфузируемую периферическую камеру (мускулатура, жир).
b. Большинство внутривенных анестетиков метаболизируется в печени (некоторые метаболиты активны) с последующей экскрецией более водорастворимых метаболитов почками.
c.
 |
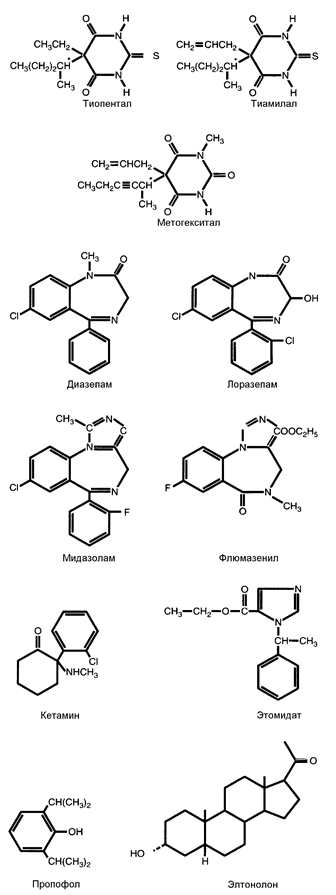
Рис. 13-1. Химическая структура неопиоидных внутривенных анестетиков.
d. Высокая концентрация устойчивого состояния в плазме достигается при длительной инфузии, когда насыщаются ферментные системы печени, и уровень выведения становится независимым от концентрации препарата (кинетика нулевого порядка).
e. Перфузионно-ограниченный клиренс описывает печеночный клиренс препаратов (этомидат, пропофол, кетамин, метогекситал, мидазолам), экскреция которых, в основном, зависит от доставки анестетика к печени (печеночный кровоток снижается при операциях в верхней части брюшной полости и при старении).
3.
 |
a. Широкий диапазон Т1/2В отражает различия в объеме распределения (Vp) и/или клиренсе.
b.
 |
4. Время полувыведения после прекращения постоянной инфузии — это соотношение времени, необходимого для снижения концентрации в плазме на 50%, и длительности инфузии (важно для определения времени пробуждения после инфузии седатиков и гипнотиков различной длительности действия) (см. гл. 47, раздел IV В).
5. Многие факторы влияют на вариабельность фармакокинетики внутривенных седативно-гипнотических препаратов у разных пациентов (табл. 13-3).
C. Фармакодинамика
1. Главным фармакологическим эффектом внутривенных анестетиков является дозозависимая депрессия ЦНС (кривая доза-ответ), проявляющаяся седацией и гипнозом (рис. 13‑3).
2. Когда в плазме достигается концентрация устойчивого состояния, можно полагать, что концентрация в плазме уравнялась с концентрацией в месте действия (рецепторы).
Уважаемый посетитель!
Чтобы распечатать файл, скачайте его (в формате Word).
Ссылка на скачивание - внизу страницы.




